non-coding regionのSNPデータを解析する
##SNPの意外な活用法
SNP(Single Nucleotide Polymorphism)の解析と言ったらはGWASの中で疾患との相関を調べたりするのに使われるものだと思っていたが、実はゲノム編集細胞のQCにも非常によく活用することができるということを最近業務の中で知った。
たとえば、SNPデータは以下のようなことに使える。
- 配列を変更したい時に天然のバリアントを利用することができる
- Single cell cloneだったものがモザイクになったら不均一集団になってしまっている
このように、GRCh38 (hg38) のリファレンス配列だけでなくSNPにまで目を向けると色々なことが分かるようになる。(実際は同僚に教えてもらったのだが・・・)
年の瀬を迎え、時間もできたことだし、予てから気になっていたDB情報を活用する方法について調べてみた。
##データベースの選定
データベースには以下のものなどがあるが、dbSNPには全てのデータが統合されている。
| データベース | データの種類 | データ量 | 更新頻度 | アクセス方法 | 主な用途 |
|---|---|---|---|---|---|
| dbSNP | SNP、indel、マイクロサテライトなど | 33億件のSNPレコード、11億件のReference SNPレコード | 約3~4か月ごと | Entrez SNP検索ツール、バッチクエリ | ゲノムワイド関連解析、集団遺伝学、進化生物学 |
| ClinVar | 遺伝的バリアントと疾患の関係 | 82.5万件以上のレコード、50万件以上のバリアント | 毎週、毎月包括的なリリース | E-utilities、Entrez Direct | バリアントの臨床的意義の解釈、医療遺伝学 |
| gnomAD | エクソン、ゲノム | 730,947のエクソームシーケンス、76,215の全ゲノムシーケンス (v4.1) | 各gnomADリリースごと | gnomADブラウザ、AWS、Microsoft Azure、Google Cloud | 希少バリアントの解析、遺伝子機能の研究 |
| 1000 Genomes Project | ゲノム | 2,504人分のゲノムデータ (Phase 3) | 更新なし | IGSRデータポータル、AWS、Ensembl、UCSC Genome Browser | 集団遺伝学、ヒト遺伝的多様性の研究 |
###dbSNPとは
dbSNPは、NCBI(National Center for Biotechnology Information)がNHGRI(National Human Genome Research Institute)と共同で開発・運営している、様々な生物種の遺伝的変異に関する情報を集約した無料の公開データベース。dbSNPは、1998年に設立され、ヒトゲノムプロジェクトで発見された多数の共通変異を収容するために作成された。dbSNPには、単一塩基置換、短い欠失・挿入多型、マイクロサテライトマーカー、レトロトランスポゾンなどの多様な分子多型が含まれている。
エントリ数が多いのは素晴らしいが、false positive rateが高いという指摘があるらしい。wikipediaでの指摘は論文での考察結果がよくまとまっているので参照されたし。
##dbSNPを使って特定の領域に含まれるSNPデータを全て取得する
dbSNPではchromosome, start position, end positionなどを指定して検索することができる。

使用可能な特殊クエリには以下のようなものがある。6[Chromosome]のように、検索後を特殊カッコの前に付けて使用する。
Gene Nameを指定したとしてもexonないしはintronが対象になるだけなので、promoter配列やそれより上流は染色体位置などを参考にしないといけない。
- [Chromosome]
- 1000[Base Position] : 2000[Base Position]
- [Gene Name]
- [Validation Status]
- [Function Class]
- [SNP ID]
- [SNP Class]
検索画面右上から検索結果をtsvで出力することも可能。出力には結構時間がかかるので気長に待つ。APIでダウンロードできるような記述をChatGPTが返してくることもあったが、未検証。
##データ分析例
出力データは以下のようになった。
#chr pos variation variant_type snp_id clinical_significance validation_status function_class gene frequency
6 187649 T>A,C,G snv 1184 by-frequency;by-alfa;by-cluster intron_variant;non_coding_transcript_variant LOC285766;LOC105374869 T:0.066677:427:1000Genomes|T:0.078473:148:HapMap|T:0.035495:104:KOREAN|T:0.071429:4:Siberian|T:0.040873:57:TOMMO|T:0.088145:23331:TOPMED|T:0.109392:3359:ALFA
6 102893 A>G snv 382895 by-frequency;by-alfa G:0.:0:ALFA
6 103136 C>A snv 462661 by-cluster
6 103505 T>C,G snv 467420 by-cluster
6 165632 A>C,G,T snv 719065 by-frequency;by-alfa;by-cluster A:0.349781:2240:1000Genomes|A:0.293186:611:HGDP_Stanford|A:0.413136:780:HapMap|G:0.430034:1260:KOREAN|G:0.411572:754:Korea1K|G:0.323529:22:Qatari|A:0.:0:SGDP_PRJ|A:0.211538:11:Siberian|G:0.453075:12803:TOMMO|A:0.272368:72093:TOPMED|A:0.19265:6060:ALFAFrequencyというカラムの値が一定値以上のもとを取ってくるなどすれば先程のfalse positive問題は回避できそう。
###データ読み込み
o1のコードだが、データの整理からしてみる。
import pandas as pd
import numpy as np
# TSVを読み込む
df = pd.read_csv("snp_result.txt", sep="\t", dtype=str)
# frequency列がNaNの場合は除外
df = df[~df["frequency"].isna()]
# frequency列が空文字や"0."のみのような場合を除外する関数を定義
def has_valid_freq(freq_str):
# freq_strが空ならFalse
if not freq_str.strip():
return False
# パイプで区切られた各エントリに少なくとも一つ数値が入っていればTrue
items = freq_str.split("|")
for item in items:
# 例: "T:0.066677:427:1000Genomes"
parts = item.split(":")
if len(parts) == 4:
allele, freq_val, count, dataset = parts
# freq_valが数値かつ0以外ならvalidとみなす
try:
val = float(freq_val)
if val > 0.0:
return True
except ValueError:
pass
return False
df = df[df["frequency"].apply(has_valid_freq)]
# frequency列を展開してtidyにする
rows = []
for idx, row in df.iterrows():
freq_entries = row["frequency"].split("|")
for entry in freq_entries:
parts = entry.split(":")
if len(parts) == 4:
allele, freq_val, count, dataset = parts
# freq_valが数値に変換できなければスキップ
try:
val = float(freq_val)
except ValueError:
continue
# 0以下はスキップ
if val <= 0.0:
continue
row_dict = {
"#chr": row["#chr"],
"pos": row["pos"],
"variation": row["variation"],
"variant_type": row["variant_type"],
"snp_id": row["snp_id"],
"clinical_significance": row["clinical_significance"],
"validation_status": row["validation_status"],
"function_class": row["function_class"],
"gene": row["gene"],
"allele": allele,
"frequency": val,
"sample_count": count,
"dataset": dataset
}
rows.append(row_dict)
tidy_df = pd.DataFrame(rows)
tidy_dfこれでtidyな使えるデータになる。
#chr pos variation variant_type snp_id clinical_significance \
0 6 187649 T>A,C,G snv 1184 NaN
1 6 187649 T>A,C,G snv 1184 NaN
2 6 187649 T>A,C,G snv 1184 NaN
3 6 187649 T>A,C,G snv 1184 NaN
4 6 187649 T>A,C,G snv 1184 NaN
5 6 187649 T>A,C,G snv 1184 NaN
6 6 187649 T>A,C,G snv 1184 NaN
7 6 165632 A>C,G,T snv 719065 NaN
8 6 165632 A>C,G,T snv 719065 NaN
9 6 165632 A>C,G,T snv 719065 NaN
validation_status \
0 by-frequency;by-alfa;by-cluster
1 by-frequency;by-alfa;by-cluster
2 by-frequency;by-alfa;by-cluster
3 by-frequency;by-alfa;by-cluster
4 by-frequency;by-alfa;by-cluster
5 by-frequency;by-alfa;by-cluster
6 by-frequency;by-alfa;by-cluster
7 by-frequency;by-alfa;by-cluster
8 by-frequency;by-alfa;by-cluster
9 by-frequency;by-alfa;by-cluster
function_class gene \
...
6 T 0.109392 3359 ALFA
7 A 0.349781 2240 1000Genomes
8 A 0.293186 611 HGDP_Stanford
9 A 0.413136 780 HapMap ###datasetの種類
特定のSNP_IDに対して様々なdatasetの測定結果が格納されているらしい。どんなdatasetがあるのかみてみる。
tidy_df['dataset'].unique()array(['1000Genomes', 'HapMap', 'KOREAN', 'Siberian', 'TOMMO', 'TOPMED',
'ALFA', 'HGDP_Stanford', 'Korea1K', 'Qatari', 'Daghestan',
'GnomAD', 'PAGE_STUDY', 'GENOME_DK', 'SGDP_PRJ', 'PRJEB36033',
'NorthernSweden', 'GoNL', 'ALSPAC'], dtype=object)経験的にこういうデータって欠損値だらけのdatasetが含まれていることが多く、省けるものもあったりする。どのdatasetに欠損が多いか確認してみる。
import polars as pl
# polars objに変換
df_tidy = pl.DataFrame(tidy_df)
# wide dataに変換 この操作はpolarsの方がやりやすい
df_freq = df_tidy.select(
[
# '#chr',
# 'pos',
# 'variation',
# 'variant_type',
'snp_id',
# 'function_class',
'frequency',
# 'sample_count',
'dataset'
]
).pivot('dataset', index='snp_id', aggregate_function='first').fill_null(0.0).filter(
(pl.col('TOMMO') > 0.0) | (pl.col('1000Genomes') > 0.0)
).head(20)
df_heat = df_freq.to_pandas().set_index('snp_id')
df_heat
# pyComplexHeatmapでplot
import matplotlib.pylab as plt
import numpy as np
plt.rcParams['figure.dpi'] = 100
plt.rcParams['savefig.dpi']=300
plt.rcParams['font.family']='sans serif' #please remove this line if font is not installed
plt.rcParams['font.sans-serif']='Arial' # please remove this line if Arial font is not installed
plt.rcParams['pdf.fonttype']=42
# sys.path.append(os.path.expanduser("~/Projects/Github/PyComplexHeatmap/"))
import PyComplexHeatmap as pch
plt.figure(figsize=(5, 5))
pch.ClusterMapPlotter(data=df_heat, col_cluster=False, row_cluster=False, show_colnames=True,
show_rownames=True)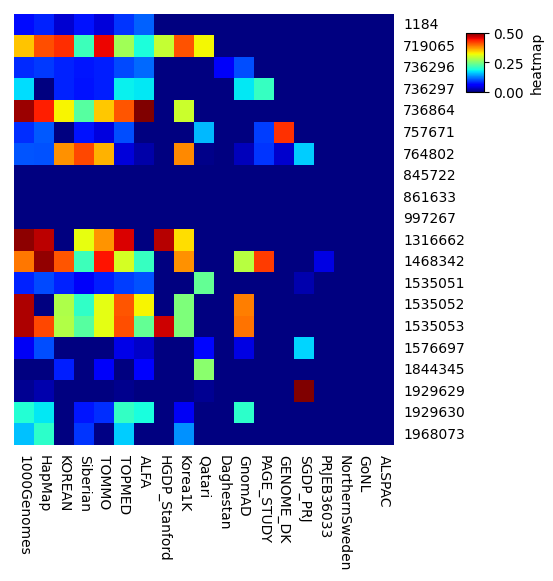
大体以下のような知見が得られた。(主観です) 今回は試しにchromosome6のごくごく一部のSNPだけを題材にしてみたが、もっと広いところを見れば異なるdatasetもあるのだと思う。用途に応じて使い分けると良さそう。
データが多いがほかのdatasetとはfrequencyの傾向が違う
- 10000Genomes
- HapMap
しっかりデータが多い
- KOREAN
- Siberian
- TOMMO
- ALFA
- Korea1K
少しデータが足りない
- HGDP_Stanford
- Qatari
- GnomAD
データ不足でスクリーニング用途では使えなそう
- Daghestan
- PAGE_STUDY
- GENOME_DS
- SGDP_PRJ
- PRJE36033
- NorthernSweden
- GoNL
- ALSPAC
-
wikipedia参照. ↩